TWOJA PRZEGLĄDARKA JEST NIEAKTUALNA.
Wykryliśmy, że używasz nieaktualnej przeglądarki, przez co nasz serwis może dla Ciebie działać niepoprawnie. Zalecamy aktualizację lub przejście na inną przeglądarkę.
Data: 18.05.2016 Kategoria: nauka/badania/innowacje

Naukowcy z Politechniki Wrocławskiej uczestniczą w międzynarodowych badaniach nad opracowaniem narzędzi bioinformatycznych do projektowania leków. Dr Adam Gonczarek i dr Paweł Świątek z Wydziału Informatyki i Zarządzania wykorzystują do tego metody uczenia maszynowego
- To duży projekt finansowany z grantu Narodowego Centrum Badań i Rozwoju, jego celem jest stworzenie komercyjnych narzędzi wspomagających laboratoria i firmy farmaceutyczne w poszukiwaniu nowych leków, ang. drug discovey – wyjaśnia dr Adam Gonczarek z Katedry Informatyki. - Będziemy pracować nad dwoma zagadnieniami, pierwsze to opracowanie kilku modułów, które mają zautomatyzować analizę eksperymentów z użyciem jądrowego rezonansu magnetycznego, w skrócie NMR-u, a drugie dotyczy tzw. screeningu wirtualnego – mówi naukowiec.
Metoda szybsza i tańsza
Projekt koordynuje firma Indata S.A. Politechnika Wrocławska jest jednym z partnerów naukowych. – Badania mają charakter interdyscyplinarny, potrzebna jest tu wiedza biologów, chemików, informatyków i specjalistów od uczenia maszynowego – wyjaśnia dr Paweł Świątek. Dodaje, że obszar, w którym specjalizuje się ich zespół, to metody uczenia głębokiego, ang. deep learning. – Mamy tu duże pole do popisu, bo to temat świeży, a badania nad zastosowaniem uczenia głębokiego do poszukiwania leków prowadzone są na świecie od ostatnich kilku lat – mówi dr Adam Gonczarek.
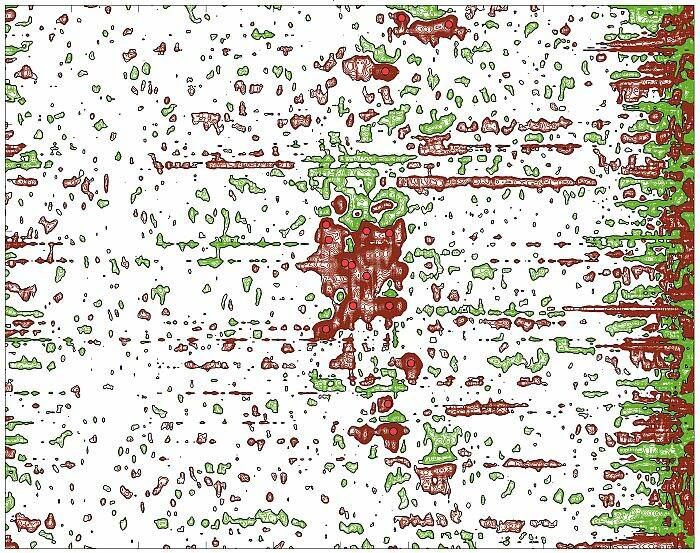
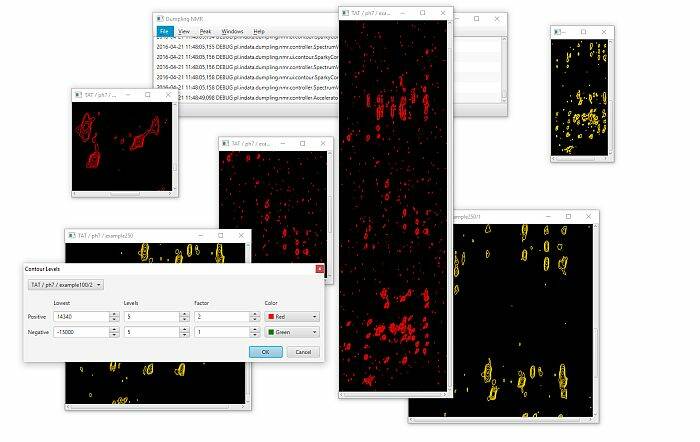
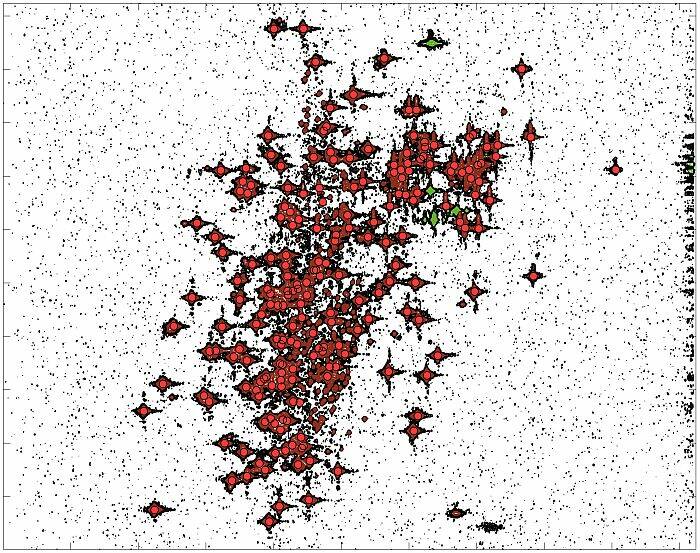
Nasi naukowcy wyjaśniają, na czym będzie polegać ich praca: - W pierwszej części chodzi o stworzenie oprogramowania do badania struktury i właściwości złożonych związków chemicznych, a w szczególności białek – tłumaczy dr Paweł Świątek. – Obecnie wygląda to tak, że badając białka przy użyciu NMR-u, naukowcy otrzymują duże zbiory wielowymiarowych obrazów. W pierwszej kolejności należy takie obrazy oznaczyć, czyli ręcznie wybrać pozytywne sygnały widmowe, a następnie przyporządkować je do odpowiednich atomów w białku. Na podstawie tak przetworzonego eksperymentu można dalej prowadzić badania o znaczeniu biologicznym, czyli analizę struktury i funkcji białek. Naszym zadaniem jest zautomatyzowanie procesu ręcznego przetwarzania danych pochodzących bezpośrednio ze spektrometru NMR. Obecnie proces ten zajmuje od kilku tygodniu do kilku miesięcy pracy. Szacujemy, że uda nam się skrócić ten czas nawet o 95 procent. Z tym wiąże się znaczne obniżenie kosztów badań oraz zwiększenie ich przepustowości – mówi dr Gonczarek.
Algorytm wspomoże badacza
Tłumaczy, że ich badania wspomogą poznanie i rekonstrukcję cząsteczek białek. - Od struktury zależy, jaki potencjalny lek będzie w stanie się związać z danym białkiem i tym samym zahamować proces chorobotwórczy. Naszym zadaniem będzie też opracowanie mechanizmu do śledzenia zmian w strukturze białka, które zachodzą w wyniku odziaływania z małocząsteczkowym związkiem chemicznym, czyli potencjalnym lekiem – wyjaśnia dr Gonczarek.
Specjaliści z PWr opracują algorytmy, która mają za zadanie wspomóc człowieka w analizie eksperymentów. Dr Świątek: - Nie będziemy w stanie całkiem wyeliminować czynnika ludzkiego, czyli udziału badacza. Metody oparte o statystyczne wnioskowanie zawsze są obarczone pewnym błędem. Dlatego, przynajmniej w części przypadków, ręczna korekta będzie potrzebna, ale i tak proces zostanie znacznie przyspieszony.
Screening wirtualny
Druga część ich badań w tym projekcie będzie dotyczyć screeningu wirtualnego (ang. virtual screening). – To metoda do komputerowego przewidywania aktywności różnych związków chemicznych z danym białkiem chorobotwórczym – wyjaśnia dr Gonczarek. - W klasycznym screeningu polega to na tym, że eksperymentalnie bada się zdolność białka do oddziaływania z setkami tysięcy małych związków chemicznych. My, wykorzystując tu uczenie maszynowe, chcemy przewidzieć, które substancje z dostępnych baz danych, mogą reagować z danym białkiem. Algorytmy uczenia maszynowego charakteryzują się tym, że najpierw muszą dostać rzeczywiste przykłady, na bazie których mogą się nauczyć, uogólniają tę wiedzę i postępują w analogiczny sposób na podobnych przypadkach – dodaje.
W Laboratorium Technologii Usługowych i Sieciowych
Naukowcy będą potrzebować do swoich działań maszyn o dużej mocy obliczeniowej. - Przetworzenie takiej ilości danych i końcowe nauczenie odpowiedniego algorytmu uczenia głębokiego to około 3-4 miesiące ciągłej pracy komputerowego klastra. - Wykorzystamy w tym celu klastry obliczeniowe naszego wydziałowego Laboratorium Technologii Usługowych i Sieciowych – mówi dr Świątek.
Badania są prowadzone we współpracy ze światowymi specjalistami w dziedzinie poszukiwania leków, m.in. z laboratorium na Politechnice Zurychskiej (ETH Zurich, dwie Nagrody Nobla z dziedziny NMR-u białek), z Universytetu Harvarda oraz z Uniwersytetu Goethego we Frankfurcie.
- Ten projekt to dla nas duże wyzwanie, dlatego cieszymy się, że udało nam się nawiązać współpracę z zespołem z Politechniki Wrocławskiej – uważa Mateusz Szuściak - dyrektor operacyjny Indata S.A.
Iwona Szajner
* Projekt badawczy "Opracowanie narzędzi bioinformatycznych do poszukiwania leków" jest współfinansowany przez Ministerstwo Nauki i Szkolnictwa Wyższe i Unię Europejską w ramach Programu Operacyjnego Inteligentny Rozwój (nr umowy POIR.01.01.01-00-1083/15).

Nasze strony internetowe i oparte na nich usługi używają informacji zapisanych w plikach cookies. Korzystając z serwisu wyrażasz zgodę na używanie plików cookies zgodnie z aktualnymi ustawieniami przeglądarki, które możesz zmienić w dowolnej chwili. Ochrona danych osobowych »